How I Found My Health “Super Unlock” After 20 Years of Research and 20,000 Genes Tested
How I Found My Health “Super Unlock” After 20 Years of Research and 20,000 Genes Tested
Each person has one to six highly unique unlocks that will only work for them, and this is how to find them.

House: I need a genetic disease.
Wilson: I’m sure you’re carrying a few.
— House MD, Season 5, Episode 6
Disclaimer: I am not a medical doctor and this is not medical advice.
There are basic things we should all be doing for good health. Eat a nutritious, largely whole-food diet that is adequate in all the macronutrients and micronutrients; maintain a healthy body composition; live an active lifestyle with healthy movement patterns and plenty of outdoor time; get outdoor sunshine in the morning and avoid artificial light at night; include exposure to fire; let our bare skin touch the earth; experience the transitions between hot and cold that nature offers; sleep well; take bites of life only as big as we can chew, but always be taking bites; nourish positive relationships with our family and friends and a positive outlook within ourselves; be mindful of avoiding excessive exposures to the toxins we may encounter; and so on.
I propose herein that most of us have an idiosyncratic portion of what we need for optimal health, and this is far more likely to be driven by the presence of one or more rare metabolic disease genes discoverable only with whole genome sequencing than it is to be driven by common polymorphisms that are accurately tested with services such as 23andMe.
For example, let us briefly consider my own health.
I had a night-and-day transformation between a neurological nightmare on a vegan diet and high-performance stability and sanity on a diet rich in organ meats. While I have known many ex-vegans, I know very few with such radical transformations as mine. I have spent twenty years wondering why this is, until recently, without success.
I have suspected there is something different about me that explains both that and why my neuromuscular health remains so sensitive to stresses:
Why did a single day of the anti-fungal terbinafine taken in 2017 give me the worst twitching I’ve ever had in my life?
Why did six weeks of it produce a twitching problem that no neurologist could diagnose but that I myself could render completely asymptomatic with supplements that I had to maintain for 4.5 years until the problem was gone?
Why did taking eggs and meat out of my diet and replacing them with shellfish for 40 days in early 2022 give me muscular weakness and trouble swallowing that disappeared as soon as I put the eggs and meat back in?
Since 2017, I have occasionally made a deep dive into rare genes and biochemistry to help clients with highly unusual, chronic, intractable health problems. And I had suspected that applying this approach to myself one day could finally explain what seemed so different about me.
Only in the last year have I moved forward to do exactly that. I have identified a rare, riboflavin-responsive mutation in the ACAD9 gene as well as an assortment of genetic assaults on the mitochondrial respiratory chain that perfectly explain everything I’ve described above. These were only discoverable through the analysis of my whole genome sequencing raw data files, and were not discoverable with the other genetic testing I had done.
Most of the past experiences with dietary changes could be explained by variations in my intake of riboflavin, pantothenic acid derivatives, and carnitine. I then leveraged this knowledge to not simply explain past problems but obtain better and better health by addressing the mutations in a targeted way.
Riboflavin supplementation alone has abolished my seasonal allergies, dramatically improved my body composition, and increased my motivation, mental health, productivity, and energy.
I am purposefully experimenting on myself to generate insights that will help others and so I am moving slowly to collect maximal data. I have a plan for the next phase of my experiments that I expect to reverse two decades of chronic discomfort from muscle tension.
As I have realized how responsive my own health is to this approach, I have applied it to more and more of my clients. Here are some of the results I have achieved with this:
One of my clients had been completely unable to have blood drawn in the fasting state and could only have it drawn while laying down or else she would faint. After addressing rare biotin-responsive mutations, she has become more fasting tolerant and just had blood drawn sitting up for the first time in her adult life. We believe she is well on her way to tolerating blood drawn in the fasting state.
One of my clients takes humira for Crohn’s disease but at the cost of neurological side effects. We identified an apparent defect in lysine metabolism. Addressing it appears to have improved her skin and energy levels, and to allow her to tolerate pushing out the monthly humira dose by two weeks without any gastrointestinal side effects. We will be seeing soon if fully addressing this issue can allow her to get off the humira entirely and not worry about neurological problems or gastrointestinal issues.
One of my clients was dealing with insomnia, anxiety, and neuropathy through benzos, SSRIs, and beta-blockers from conventional doctors, through hormonal treatments from functional medicine doctors that worked until they didn’t, and had tried supplementation strategies using phosphatidylserine and inositol that made him tremendously worse. This client came to me for my advice on methylation, but after identifying a problem in complex I of the mitochondrial respiratory chain we identified restricting simple carbs as a major way to achieve the first reduction of symptoms he had experienced in years.
One of my clients with longstanding insomnia responsive only to sleep drugs has achieved months of 7-8 hours of sleep per night through glutathione infusions after we identified an apparent defect in glutathione synthesis.
One of the great drawbacks of applying this approach gradually is the the massive delays in achieving these results. For example, in Energy Metabolism Governs Everything, I posited that a respiratory chain disorder was causing one of my clients to require much higher than normal doses of vitamin A and zinc, was causing paradoxical coexistence of vitamin A deficiency and toxicity symptoms, and was driving her apparent autoimmune condition.
It later turned out that I was right on the money. This client had a complex III disorder traceable to six homozygous mutations in the MT-CYB gene. However, I had started working with this client in March of 2022 and formed this hypothesis in April of 2023. The time it took her to get the necessary labs run led me to only finally develop an action plan for her this past month.
What caused this delay? The fact that I 1) only pursued rare defects in energy metabolism when all other explanations had been exhausted, and 2) left her on her own to find doctors to sign off on her labs.
Recent research I will review below has shown that everyone has one or two such defects. I show my math below to suggest that each person actually has between one and six nutritionally actionable defects in energy metabolism. This does not mean that everyone is walking around with one to six serious illnesses. It means that everyone has one to six idiosyncrasies of energy metabolism and addressing them for some people will resolve chronic health problems and in other people will simply make them even healthier than they already are.
For some, it is solution to a longstanding problem.
For others, it is the highest-return unlock for maximal wellness, performance, and longevity.
For this reason, I created a program that will start by getting all of the relevant labs ordered for you and will systematically identify the highest-return idiosyncrasies from the beginning, giving you a highly individualized nutrition and supplement plan to address them within 12-14 weeks. You can sign up for a call at bioopthealth.com to see if my new Biochemical Optimization Program is right for you.
This Article is Only Free for 48 Hours
This article is only free for the first 48 hours, until 5:00 PM Eastern on the evening of Saturday September 23, 2023. After that, it is reserved for Masterpass members. Learn more about the Masterpass here.
Or, sign up here:
In This Article
Rare Metabolic Disease Genes Require Whole Genome Sequencing to Analyze
The High-Hanging-Fruit Unlock for Optimizing Wellness, Performance, and Longevity
Debugging the OS
The evidence we will look at below suggests each person has at least “carrier” status for one to twelve mutations in metabolism.
Roughly half of these are defects in energy metabolism that are actionable deserve the overwhelming attention.
Energy metabolism is the operating system. For short, the OS.
Everything else — your skin, your hair, your fatigue, your athletic power, your brain power, your cardiovascular health, your propensity toward cancer, your immune system — is an app that runs on the OS.
Debugging the OS is the next big step for everyone after the low-hanging fruit is brought into order. It is their unique set of unlocks for wellness, performance, and longevity.
Debugging the OS means identifying those one to six defective genes in each person that are 1) rare and severe, 2) fundamentally involved in energy metabolism, and 3) highly actionable.
The solutions will often be a shift in the proportions of protein, fat, or carbohydrate, avoiding or utilizing high doses of specific vitamins or minerals, and in some cases using custom-designed amino acid mixes or certain specific types of fat.
Most of conventional and functional medicine is focused on treating the glitching apps. Conventional medicine treats the glitching apps with drugs in the service of pharmaceutical and health insurance companies. Functional medicine tries to do better and tries to pursue better values, but winds up treating the glitching apps with bags of supplements.
Most nutrigenomics is focused on the large number of common polymorphisms and not the small number of high-impact unlocks. As a result, it does little beyond produce a massive tutorial on the details of the code that underlies the function of each of your apps.
Even the whole genome sequencing companies give you dozens of reports that each cover dozens of genes with no coherent system for prioritizing them or filtering them by how actionable they are.
Debugging the OS is how to resolve confusing and intractable problems.
Debugging the OS is how to bring performance to the next level when one has already done everything else right.
Debugging the OS is how to take someone whose genes say they die early, debug the most important of them, defy the morbid destiny, and earn them centenarian status.
Debugging the OS is how to find the one to six highest-impact unlocks for maximal wellness, performance, and longevity.
Keep reading if you want to understand the science that inspired the program.
This Is Not Conventional Wisdom
This strongly diverges from conventional wisdom.
The conventional wisdom is that only a portion of those with two copies of a single defective gene are impacted by the defect, as a result of environmental components and modifying genes that reduce the “penetrance” of most defects to less than 100%, which means fewer than 100% of people with the genes get the disease, and with exceedingly rarer impacts from other types of inheritance, such as dominant genes or X-linked genes.
However:
Rarely, but often enough to take note, full-blown inborn errors of metabolism due to a traditionally homozygous inheritance occur, and many if not most go undiagnosed.
Far more common than that, two heterozygous mutations in closely related pathways produce a disease of the same full-blown magnitude, yet these are almost never ever diagnosed. This is called synergistic heterozygosity.
Far, far, far more common than that, impacting virtually everyone, there are one to six heterozygous defects that do not combine to produce a full-blown disease but do constitute idiosyncratic bottlenecks in metabolism that do impact health, usually in a subtler way that is often delayed in onset until the fourth decade of life or later. A very well-established example of this is heterozygous familial hypercholesterolemia.
In all three cases, the defective gene can be seen as a bug in the operating system, and if it has actionable implications, it can be debugged. Debugging the OS may help mitigate severe problems in some cases, and in most cases may would be critical to optimizing wellness, performance, and longevity.
When people experience seemingly magical cures to health problems such as psychiatric diseases, dysautonomia, fatigue, or intractable gut or skin problems with something like a ketogenic diet, 2000 milligrams a day of thiamin, or some other extreme change in macronutrients or micronutrients, they are debugging their OS by accident.
The problem is that people share their experience but do not understand the idiosyncratic nature underlying their response. Others copy them, hoping for the same results, and instead have the complete opposite experience, sometimes causing months or years of a difficult path to recovery.
Just in the month before writing this, I had several consulting clients who had dysautonomia get much worse with thiamin or with a ketogenic diet, and one who had a honeymoon period with thiamin followed by a crash.
One of them had clear evidence of a fatty acid oxidation disorder, and had gone from bedridden to getting out of bed regularly simply by modestly increasing carbs and cutting out high-dose thiamin.
As I wrote about in How to Interpret Ketone Ratios and the Lactate-to-Pyruvate Ratio, getting worse on thiamin is a huge red flag for a respiratory chain disorder or an imbalance with a nutrient specifically involved in the respiratory chain, such as copper. Getting worse on a ketogenic diet (beyond the normal “keto flu” or a slump in athletic performance that can easily be attributable to glycogen depletion) is a big red flag for a disorder in fatty acid oxidation or ketone utilization, or a deficiency in a nutrient disproportionately used in these pathways, such as riboflavin.
If we can be more precise in our understanding of each person’s unique glitches in their OS, we can minimize the harm of making the opposite dietary change as needed, and we can broaden the experience of having amazing results.
Bugs in the OS will turn out to be critical to the resolution of confusing and intractable health problems and will turn out to be the most powerful unlocks of general wellness, performance, and longevity.
Here are examples of health problems that debugging the OS will help solve:
Most of the NIH Rare Diseases program.
All epilepsy with no distinct physical injury, structural anomaly, or biochemical insult identified as the cause.
Everyone on the autism spectrum.
Early-onset cases of the neurological diseases of aging (young-onset Parkinson’s, for example).
Most people with chronic fatigue syndrome/myalgic encephalomyelitis.
Most people passed around from doctor to doctor with no one who can figure out what is wrong with them.
Most people whose doctors insist their symptoms are all in their head or are results of anxiety and hypochondriasis who are themselves convinced otherwise.
Most people with genuine psychiatric diseases.
Here are examples of the massive benefits that can be obtained by debugging the OS even in someone without confusing health problems:
Helping an athlete advance from silver to gold.
Take someone from a family with relatively poor longevity and help them reach centenarian status.
The logic is as follows: if you are doing everything right, but the biggest idiosyncratic glitch in your own personal energy metabolism is that half of your carbohydrate metabolism doesn’t work unless you are on 500 to 2000 milligrams of thiamin per day, how could knowing this and acting on it not be the biggest possible unleash of your fullest biochemical potential?
Rare Metabolic Disease Genes: Defining Exactly What I Mean
By rare metabolic disease genes I am talking about the genes for inborn errors of metabolism, rare “single-gene” (in appearance, not in reality) defects in specific components of biochemical pathways. There are approximately 1,450 of these.
Within these, when facing combinations of more than one, those affecting the utilization of energy dwarf the importance of all others, because Energy Metabolism Governs Everything.
Energy metabolism is the operating system (OS).
Everything else is an app.
None of your apps will work correctly when the OS is glitching.
Step 1 then is always to debug the OS.
Within the genes impacting energy metabolism, I propose that those impacting the utilization of nutrients involved in the utilization of energy are the most nutritionally actionable. Generally speaking, things that are nutritionally actionable should be targeted before anything that is actionable only with “orphan drugs” and other experimental pharmaceutical treatments simply from a risk/reward standpoint.
This comprises 323 disorders impacting the infrastructure cellular energy production, 226 directly impacting the utilization of fat, protein, or carbohydrate, and 108 directly impacting the utilization of vitamins and minerals. Thus, 657 disorders fall clearly into the category I am most concerned with.
However, this is an underestimate because some disorders are classified elsewhere but nevertheless have fundamental impacts on energy metabolism or are fundamentally nutritionally actionable. For example, many of the 118 disorders impacting metabolic cell signaling impact the regulation of energy metabolism, such as a defect in insulin signaling. Or, many of the 327 disorders of complex molecule and organelle metabolism impact energy-burning organelles like mitochondria and peroxisomes. Many of the 172 disorders in the metabolism of heterocyclic compounds have impacts on energy metabolism and have nutritional consequences, such as the disorders of the synthesis of the iron-containing protein heme. Many of the 177 disorders impacting lipid metabolism and transport are nutritionally actionable, such as the supply of cholesterol to someone with a defect in cholesterol synthesis.
Thus, roughly 700-800 of these disorders fall into the category of fundamentally impacting energy metabolism with actionable nutritional strategies.
How Common Are These?
A recent analysis of nearly one million people provides an estimate. They limited one analysis to genes with frequencies less than 0.1% that have been reported to the NIH’s ClinVar database as possibly pathogenic by two or more submitters, without any conflict in the interpretation. They found that the average person has 1.6 such mutations.
This is an underestimate because it is uses whole exome sequencing, which looks at 2% of the genome, rather than whole genome sequencing, which looks at 90% of the genome. Whole genome sequencing has about 14% better diagnositic utility than whole exome sequencing even when the scope of “diagnosis” is limited to the conventional approach. This, conservatively, suggests the average number of such mutations per person is closer to 1.8.
Further, ClinVar is a database built on anecdotes with rarely a clear case for or against causality. The Human Gene Mutation Database (HGMD) has 2.3 times as many pathogenic interpretations of genes as Clinvar. The rare exome variant ensemble learner score (REVEL Score) uses the aggregate scores of 13 different tools used to predict the likelihood a mutation is pathogenic, and it identifies 2.9 times as many likely pathogenic genes as the HGMD.
Since there is no rational basis for selecting a “gold standard” reference point, since everything is a choice between computer modeling and anecdote, I would use these various estimates to establish a range of 1.80-12.2 rare disease genes per person.
If roughly 750 out of 1450 inborn errors of metabolism are nutritionally actionable disorders of energy metabolism, then the average person has between one and six such mutations.
These are the one to six highly unique unlocks in each person that will bring their own wellness, performance, and longevity to the next level.
These are the one to six bugs that need to be cleaned from the OS.
Conventional Wisdom Calls This “Carrier” Status
Most, though not all, rare metabolic diseases are inherited through an autosomal recessive pattern. This means that two copies of the defective gene, one from the father and one from the mother, are needed for the disease to manifest. Those who have only one copy are “carriers.”
The word “carrier” is used because we “carry” a risk of a recessive disease that we might pass along to our offspring but we do not ourselves suffer from the disease.
This is how the National Human Genome Research Institute describes it:
A carrier, as related to genetics, is an individual who “carries” and can pass on to its offspring a genomic variant (allele) associated with a disease (or trait) that is inherited in an autosomal recessive or sex-linked manner, and who does not show symptoms of that disease (or features of that trait). The carrier has inherited the variant allele from one parent and a normal allele from the other parent. Any offspring of carriers is at risk of inheriting a variant allele from their parents, which would result in that child having the disease (or trait).
Wilson’s comment to House quoted at the beginning reflects the fact that most people carry a few of these diseases. This is why incest is so terrible: most of us carry recessive diseases and if we mated with our close family we would be likely to mix genes for the same recessive disease and generate children homozygous for that disease.
The Limitation of the Conventional Wisdom
This definition of “carrier” sees “disease” through the binary diagnostic medical paradigm, which imposes a reality distortion filter onto continuous variables in order to increase the utility of the data toward triaging between choices like “to treat or not to treat.” Such triaging is, in the modern context, largely in the service of pharmaceutical and health insurance companies.
This doesn’t change the fact that being heterozygous for a biochemical defect leads to having that defect operate at approximately 50% magnitude within your body.
The cell can always compensate for a defect in a pathway by ramping up pathways that get around it, which is why it may not necessarily result in any symptoms. For example, a drop in MTHFR activity can be circumvented by supplying choline or betaine to an alternative enzyme that does largely the same thing.
There are also numerous compensations that could change that number to something other than exactly 50%:
The cell can sometimes turn up the expression of the good copy of the gene to a greater level than normal and decrease the expression of the defective gene.
In X-linked diseases in females, the cell can selectively inactivate the X chromosome carrying the defect.
Because each cell has many mitochondria and they do not divide with the cell evenly, mitochondria can exhibit heteroplasmy. This means some mitochondria have different genetics than others. Mitochondria usually but not always descend exclusively from the mother. Mutations can arise within the maternal line that do not impact all of the mother’s mitochondria, and in at least some cases paternal mitochondria can be inherited alongside those from the maternal line. With heteroplasmy, if the good mitochondria duplicate more and faster than the bad mitochondria, the proportion of healthy genes can be enriched.
If a genetic defect occurred after fertilization — or, conceivably, if cells could tap into the DNA repair process to take the good gene from one parent and edit the bad gene from the other parent — some cells in a person can have a different genome than other cells, which is called mosaicism.
Despite all this, usually individuals with “carrier status” have, at the biochemical level, 50% of the defect found in the disease, at least when using simple measurements such as the activity of the relevant enzymes in blood cells. Thus, while it may not be causing a “disease,” it is still impacting the person’s metabolism. And thus, it is probably still impacting their health in some way, whether that is making it harder to heal, presenting an idiosyncratic limiting factor to optimizing their personal performance or longevity, or manifesting a subtler shadow of the actual disease later in life.
Heterozygosity For These Is the Key to Longevity
The most common impact of heterozygosity for rare metabolic disease genes is that certain aspects of the disease become delayed by decades into the fourth decade of life or later, making them the unique limiter of the carrier’s longevity.
Heterozygous Familial Hypercholesterolemia
Let’s consider the well-established example of heterozygous familial hypercholesterolemia (FH).
Homozygous FH is a very rare disease, numbering one in a million. The traditional fate of an individual born with it was to die of a heart attack in infancy. Only in the last couple of decades has plasma aphaeresis — which removes lipoproteins from the blood — allowed individuals born with homozygous FH to reach childbearing age.
As is typical for most genetic diseases, those who are heterozygous have 50% of the biochemical defect. Only because mainstream medicine became completely obsessed with understanding the relationship between cholesterol levels and heart disease has it become well established that heterozygotes — about 1 in 300 people — have 8-10 times the normal age-adjusted risk of heart disease. If we look at the survival curves, we can see how the disease is brought forward in time:
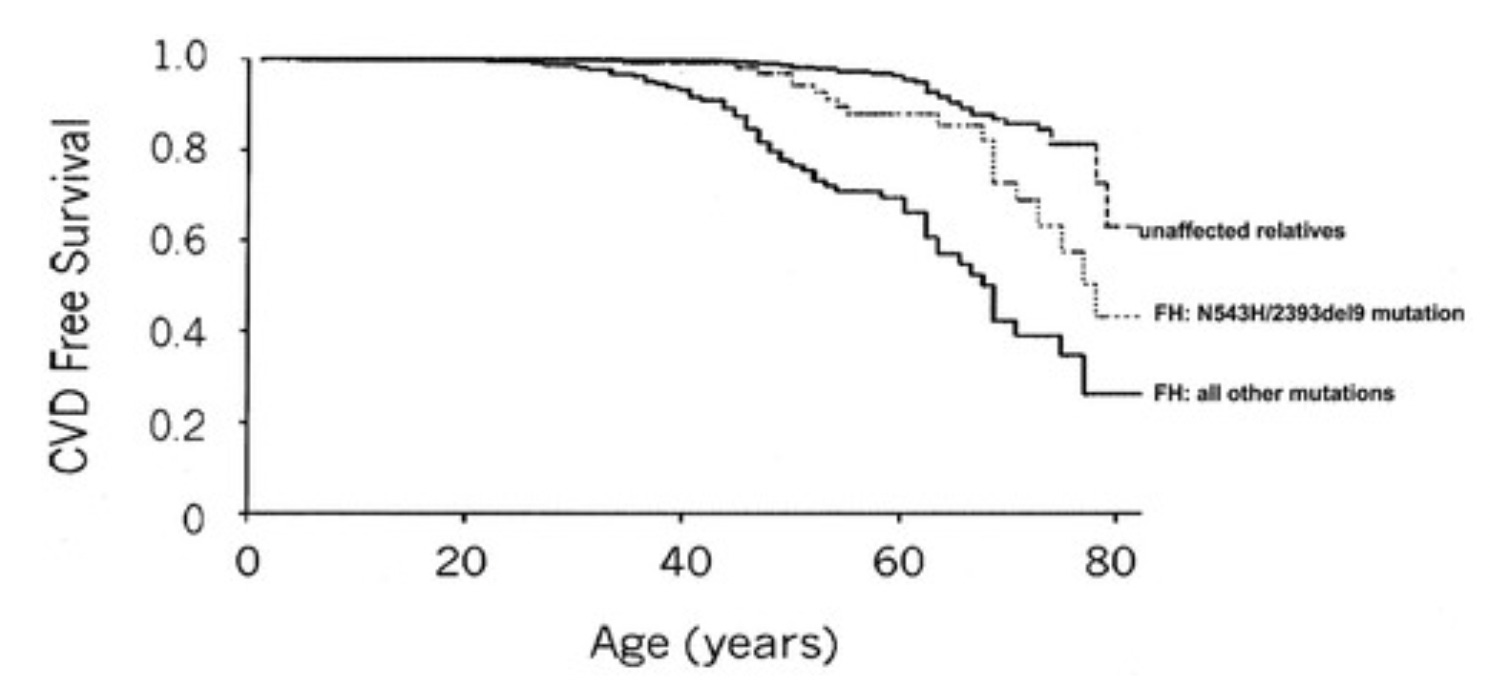
Thus, the homozygous disease is, without medical treatment, nearly a guaranteed death in infancy. The heterozygous condition is fully expressed, but it manifests not as a guarantee at any age, but rather as an increased risk of heart disease beginning around age 35, with the maximal increase of risk compared to unaffected relatives being close to age 60.
Heterozygous familial hypercholesterolemia is at the level of the cardiovascular health app, and is not a defect in the OS.
However, it is a defect in the import of LDL into the cell, which depends on energy. Specifically, import of LDL and other such molecules into a cell in this way requires the protein dynamin, which uses energy from GTP to fuel movement. Energy from GTP is exchangeable with energy from ATP via the enzyme nucleoside diphosphate kinase, so LDL import is completely dependent on mitochondrial generation of ATP.
Thus, while heterozygous familial hypercholesterolemia can be treated at the app level with a low-fat, low-cholesterol diet or a statin, the average person with this problem will also have one to six unique bottlenecks in ATP production, and debugging the OS would help them tolerate fat and cholesterol better while removing early heart disease as the primary limiter of their longevity.
The defective app determines how the energy deficiency manifests — cardiovascular disease in this case — but the bugs in the OS determine whether such a deficiency manifests.
Debugging the OS gets to the heart of the matter.
Heterozygous Biotinidase Deficiency
Now, let us take an issue that has been largely ignored by mainstream medicine rather than obsessed over: biotinidase deficiency.
The prevalence of the homozygous disease is one in 60,000, but the prevalence of “carrier” status is one in 123. By both metrics, it is far more common than familial hypercholesterolemia. Newborn screening for this disorder began in the mid-1980s, so even if focus had been put on prospectively following “carriers,” we would only have been seeing the first 35-year-olds pop up in the last couple of years. However, the fact that even many people with the enzymatic imprint of the actual “disease” were asymptomatic has led to there being no attention invested toward carriers at all.
Yet simple math and simple logic say that this is a deficiency of biotin recycling, and if one runs a lifelong deficit of biotin recycling that is smaller than what causes disease in infancy, childhood, or adolescence, it should manifest as an increase in the same problem that takes decades longer to manifest.
In other words, the same principle should be true for biotinidase deficiency as is true for the more severe and far less common disease, familial hypercholesterolemia.
We have one case report suggesting this is indeed true. A 38-year-old mother of a child born with biotinidase deficiency started developing chronic vaginal yeast infections that did not respond to treatment. Since her child had been caught by newborn screening, she knew that she and the child’s father were both heterozygous. She called up the genetic counselor and asked if biotin could help. The counselor said, I don’t know, try it. She tried it, and the vaginal yeast infections disappeared.
In this case, the high-dose biotin is debugging the OS.
It is fixing energy metabolism through biotin’s role in breaking down certain amino acids and fatty acids for energy, activating the citric acid cycle, and driving energy into the synthesis of lipids needed for mucosal barriers.
Her vaginal immune system is the app.
It runs on energy, and it depends on the energy-intensive creation of structural defenses built into the mucosa that makes its job easier.
By the sheer accident of having a homozygous baby and through thinking outside of the box rather than following the conventions of medicine, she debugged her OS with biotin and that normalized the defective app.
Heterozygous Wilson’s Disease
Wilson’s disease is a defect in copper transport that can lead to low serum copper but harmful copper accumulation in specific organs, such as the liver and brain. One in 30,000 people are diagnosed with the disease, homozygosity for disease-causing mutations is one in 7,026, and heterozygosity in the United Kingdom is as high as 2.4-5.6%. The lower number is for variants with the strongest evidence for pathogenicity.
Whether the discrepancy between diagnosed disease and homozygosity is due to low penetrance (few with the genes get the disease) or low diagnostic rates (few with the disease are diagnosed) is unclear. I would point out, however, that whether someone “gets Wilson’s” should be extremely dependent on their ATP levels, since Wilson’s is a defect in the ATP7B gene, so named because it codes for a copper-transporting ATPase that directly uses the energy of magnesium-ATP to transport copper. Thus, I would surmise that this is underdiagnosed like all the other inborn errors of metabolism but that it also has very low penetrance since it may require a perfect storm of ATP-depleting factors from other causes to manifest.
Up to 35% of carriers for Wilson’s show low ceruloplasmin or serum copper, and the majority of heterozygotes show brain imaging signs consistent with Wilson’s, but of a much lower severity. There is no direct evidence of an age-delayed decline in brain health, but I would think that a slower accumulation of free copper with time would lead to a slower type of the same damage. I would also think that age-related declines in energy metabolism would lead to greater and greater biochemical disruption of copper transport, making the rate of copper deposition start to “catch up” to diagnosable Wilson’s with age.
It has been hypothesized that heterozygosity for Wilson’s drives early-onset Parkinson’s.
Unlike biotinidase deficiency, this cannot be solved with copper supplementation. In fact, Wilson’s is typically treated with chelation, because the harms of copper deposition in certain tissues outweigh the harms of copper deficiency in others.
However, the analog of biotin supplementation for biotinidase deficiency would, for heterozygous Wilson’s, be improving ATP levels.
In this case, copper transport is an app, but it is an app that is needed for generating updates to the OS.
In more literal terms, copper transport is needed for energy metabolism because copper plays a unique role in cytochrome C oxidase, which allows the utilization of oxygen to make ATP.
However, Wilson’s disease is not highly actionable. It is treated with chelation, which is nothing but a tradeoff — trading deficient copper in some tissues to reach normal status in others rather than having adequate copper in the former tissues with toxic levels in the latter.
It is the other several bugs in the OS that impact ATP production in an actionable way that should be focused on. This will raise the level of ATP and improve the function of copper transport rather than simply engaging in a tradeoff to try to avoid the problem.
Synergistic Heterozygosity
As a result of the magnesium-ATP cofactor for the Wilson’s enzyme, the likely tens or hundreds of millions of Wilson’s “carriers” would be an excellent population to look for the operation of synergistic heterozygosity.
This is a deeply underappreciated principle that I first learned about in 2017 from a single sentence in the 6th edition of the Saudubray textbook:
It has also been suggested that symptoms, such as myopathy, may occur in individuals who are heterozygous for more than one defect of fatty acid oxidation or related pathways this proposal has been termed ‘synergistic heterozygosity’.
Synergistic heterozygosity was first proposed by Jerry Vockley and colleagues in 2000, and was recently updated by Vockley and colleagues in 2019. In this concept, heterozygous “carrier” status for multiple genes in one pathway or in closely related pathways can synergize to produce a full-blown disease that has no precise enzymatic diagnosis. This would be distinguished from compound heterozygosity, which refers to two different defects present in the same gene. It would also be distinguished from modifying genes, which have often small and numerous effects in determining the course of the disease or perhaps silencing it. In synergistic heterozygosity, a small number of genes have largely equal contributions in causing a disorder that each could not cause on their own.
Here are three examples:
A 17-year-old male was healthy until he started feeling fatigued at the start of football practice. A few weeks later he had flu-like symptoms, and then began suffering rounds of muscle cramps, vomiting, and loss of muscle protein into his urine in response to playing football. Full screening for inborn errors in energy metabolism revealed he was a heterozygous for deficiencies in complex I of the respiratory chain, CPT2 — an enzyme that helps bring fatty acids into mitochondria to burn them for energy — and myoadenylate deaminase, which breaks down AMP, the low-energy form of ATP. These appeared to combine to give him a level of myopathy that could be produced from homozygosity for one of those genes.
A 25-year-old woman presented with life-long exercise intolerance and had been complaining of muscle pain, fatigue, and swelling in her arms and thighs that made it difficult for her to climb stairs or go shopping. She was diagnosed with McArdle’s disease on the basis of elevated glycogen content and low activity of an enzyme needed to break it down. Several years later she could no longer write or walk, developed chronic fatigue setting in within 3-5 minutes of muscle use, and her muscles became progressively more painful and swollen. It turned out she was heterozygous for McArdle’s disease as well as CPT2 deficiency.
Three young or infant subjects had apparent glycogen storage diseases, presenting with such signs as enlarged livers, poor muscle tone, failure to thrive, respiratory distress, and seizures, along with hypoglycemia, acidosis, and elevated ketones. None of them had homozygosity for any one enzyme, but all of them had heterozygosity for more than one gene in glycogen metabolism. They all responded well to typical treatment, which is usually high protein (3-4 grams per kilogram bodyweight) with a carbohydrate supply such as cornstarch optimized to continuously supply glucose at a rate that meets the body’s needs without requiring storage.
Synergistic Heterozygosity Is Far More Common Than Traditional Homozygosity
Basic, simple math shows that if synergistic heterozygosity is possible, it has to be vastly more common than homozygosity for a single gene, yet incredibly harder to diagnose.
It is hard to predict a priori how many genes could combine to produce synergistically deleterious results, so let us start by taking the example of the three documented glycogen storage disease cases and isolate our view to the apparent combinations.
Figure 3 from the paper maps out the combinations within the context of the relevant biochemical pathway:
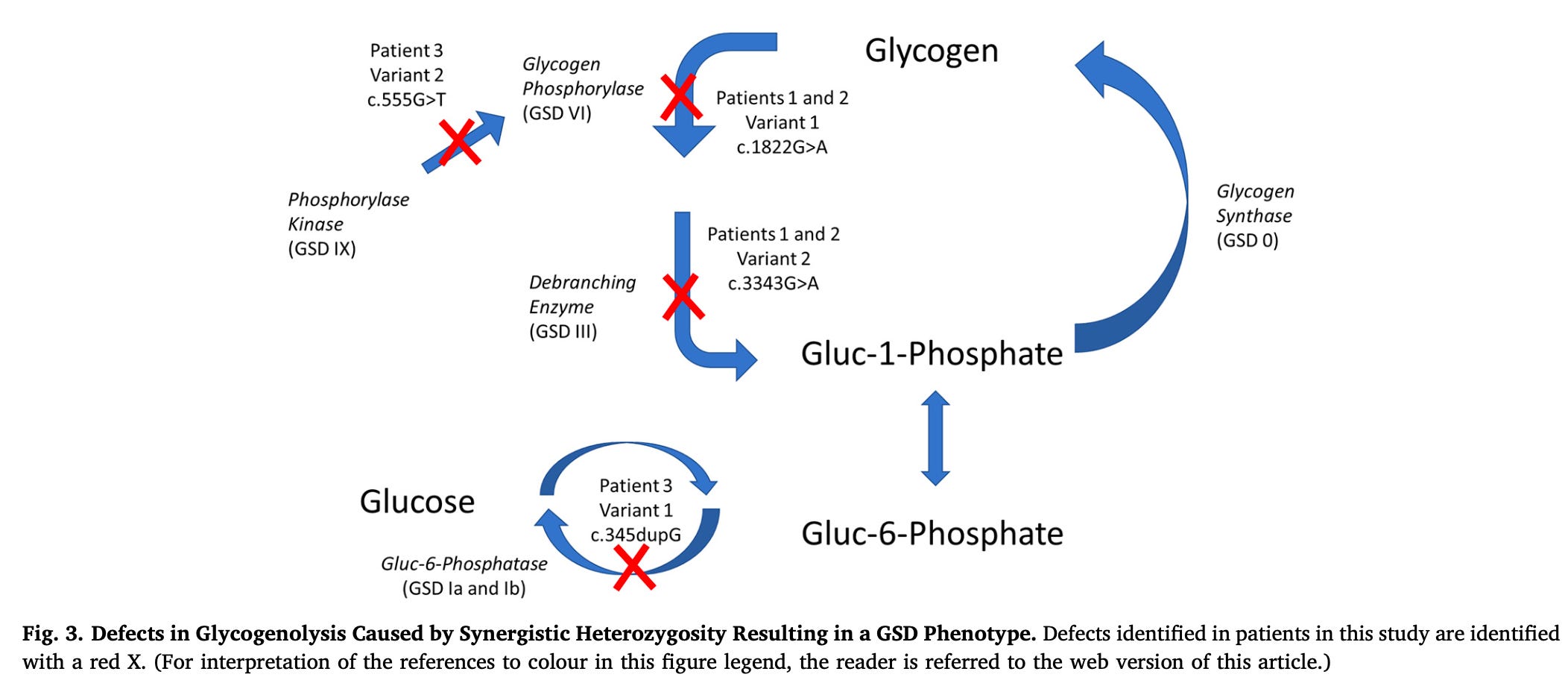
Patients 1 and 2 had successive combinations in the breakdown of glycogen to glucose-1 phosphate. From these two patients, one could argue that the defects only combine if they are very closely linked in two successive enzymes.
However, patient 3 had one mutation in the enzyme that activates the first enzyme that was defective in patients 1 and 2, and a second mutation in glucose 6-phosphatase, which allows the liver to release free glucose from glycogen breakdown into the blood.
The distance of the mutations in patient 3 creates a 4-enzyme sequence of known defects where synergistic heterozygosity can yield a full-blown disorder.
While not shown with certainty from these three cases, this suggests that any two of these combinations could result in full-blown disease.
Disregarding the incidence and diversity of these mutations and instead focusing on the basic principle involved, let’s look at whether homozygosity or synergistic heterozygosity should be more common:
For each enzyme, each person has a maternal allele and a paternal allele. Homozygosity requires both alleles to bear the same defect. There is thus one opportunity for homozygosity for each of the four enzymes, and four such opportunities.
Synergistic heterozygosity means that each one enzyme could synergize with any of the other three to produce a defective outcome. Thus, there are 16 total opportunities, but four of them are traditional homozygosity. So, the concept of synergistic heterozygosity adds 12 opportunities for a defect where there had only been four.
On this very limited basis, we could expect 3 cases of synergistic heterozygosity for every 1 case of traditional homozygosity.
More likely, the glycogen storage case underestimates the potential. For example, the 25-year-old woman with lifelong exercise intolerance had mutations in totally different pathways of energy metabolism: CPT2 brings fatty acids into the mitochondria for beta-oxidation, and McArdle’s disease is a defect in myophosphorylase, the version of glycogen phosphorylase in skeletal muscle. Fatty acids are used by the liver to generate the ATP needed for gluconeogenesis, and are used by skeletal muscle to fuel low- and moderate-intensity activity. This would have compromised the ability of her muscles to burn fatty acids and deprive them of glucose both by inhibiting the breakdown of muscular glycogen and by inhibiting the release of glucose from the liver that the skeletal muscle could take up during exercise.
If such distant pathway combinations can produce full-blown disorders, than the prevalence of synergistic heterozygosity has to be far, far more than three times as common as traditional homozygosity.
For example, chapter 12 of the Saudubray textbook lists 16 targets of defects in fatty acid oxidation and 5 closely related defects in riboflavin metabolism. Chapter 5 lists 25 genetic defects in glycogen metabolism. If these 46 targets could all interact in synergistic heterozygosity, the number of additional disorders that could be added to the first 46 would be over 2,000, leading 98% of the potential disorders to be undiscovered interactions.
Synergistic Heterozygosity Is Almost Entirely Undiagnosed
The symptomatic presentation of one of these disorders can be very different for early-onset and late-onset versions, and is usually highly heterogeneous anyway. For synergistic heterozygosity, symptoms are likely to blend into mixed presentations as well as to generate new symptoms entirely.
Thus, they are likely to be undiagnosed because synergistic heterozygosity is not recognized, because it results from the combination of two or more partial defects that each on their own are categorized as “normal,” and because the presentations are going to look unique, rather than matching any specific disorder.
This difficulty in diagnosis reinforces the failure of the concept to take hold in the field.
A pubmed search for “synergistic heterozygosity” filtered by “case reports” yields four results, including Vockley’s 2000 paper, which contained a series of 8 patients. Vockley’s 2019 paper added the three cases of glycogen storage diseases. A Google Scholar search for papers that cite Vockley’s 2019 paper and have “synergistic heterozygosity” in the title yields 5 results, none of which add additional cases.
Thus, there appear to be approximately 15 cases published explicitly evoking this concept, and there are almost 100 times as many unique inborn errors of metabolism identified, each with many cases diagnosed.
A Google Scholar search for “synergistic heterozygosity” yields 437 results, while “inborn errors of metabolism” yields 69,700 results. This suggests that about 1 in every 159 papers about inborn errors of metabolism mentions the concept.
I have the 6th edition of the Saudubray textbook, published in 2016, in hardcover, and the 7th edition, published in 2022, in Kindle. Both contain a single sentence referencing this concept in the “Disorders of Mitochondrial Fatty Acid Oxidation & Riboflavin Metabolism” chapter, which I had quoted above and quote again here:
It has also been suggested that symptoms, such as myopathy, may occur in individuals who are heterozygous for more than one defect of fatty acid oxidation or related pathways this proposal has been termed ‘synergistic heterozygosity’.
The sentence remains completely unaltered between both editions. In each case, Vockley’s 2000 paper is cited, and the 2022 edition of the textbook does not update this by citing his 2019 paper.
This concept, then, appears to have gotten very little attention in the field.
It follows that the traditional approach to diagnosing inborn errors of metabolism has likely only uncovered the tip of the iceberg, and that there are likely at least 50 times as many cases of full-blown diseases that go undiagnosed because they do not fit the traditional definition of a single defect in a single enzyme.
And why wouldn’t they be diagnosed?
Let’s take the three glycogen storage disease cases as an example. Each patient had partial defects in two enzymes. That means each enzyme is “normal” by the conventional definition, since they only “carry” these diseases and do not suffer from them. Assays of enzymatic activity and mutation analysis would each on their own and both in combination fail to diagnose all three patients.
The mutations relate to glycogen storage diseases types Ib/c, III, VI, and IXb. According to the “The Glycogen Storage Diseases and Related Disorders” chapter of the Saudubray textbook, this is how these four diseases are diagnosed:
Type I was formerly diagnosed by enzymatic activity in liver biopsy, and is now diagnosed by mutation analysis instead so that liver biopsy can be avoided.
Type III is diagnosed with enzymatic activity in white blood cells or with mutation analysis.
Type VI is diagnosed with enzymatic activity in liver or blood cells, which has a high false negative rate, or with mutation analysis.
Type IX is diagnosed with enzymatic activity in liver or blood cells, but this “may be difficult to interpret” and “diagnosis is best achieved by mutation analysis using a DNA panel.”
Thus, the three glycogen storage disease cases in the Vockley 2019 paper could never be diagnosed using conventional means.
All three were diagnosed based on clinical suspicion driven by fasting intolerance, unexpectedly high ketones and low glucose in the fasting state, and mildly enlarged livers, and by “treat it and see if it works” House MD-style diagnosis.
The list of major presenting signs and symptoms is broader than what could be used to suspect such a disorder:
Patient one was born premature and presented with respiratory distress at 34 weeks of age, mild motor developmental delay, an enlarged liver, and failure to thrive.
Patient 2 had an enlarged liver and failure to thrive.
Patient 3 was short in stature, had poor muscle tone, sensory disturbances, and seizures.
These are very non-specific signs.
It was Vockley’s expertise and instinctive clinical skill that saw through the variable presentation to test fasting tolerance and fuel molecule concentrations in the fasting state and thereby find the common thread.
But far more than that, it was Vockley’s unique conceptualization of synergistic heterozygosity and his willingness to be the lone voice crying in the wilderness that synergistic heterozygosity exists that stopped him from what doing what most other practitioners would do and saying “enzymatic activity and mutation analysis shows you do not have a specific disorder so I cannot help you.”
Now, if it is the case that there are at least 50 times as many cases of full-blown disorders due to synergistic heterozygosity than to traditional homozygosity and they are virtually all walking around without a diagnosis, who are they?
As I wrote at the outset, these are likely many of the unsolvable cases that get submitted to the NIH Rare Diseases program. It makes perfect sense. Every typical diagnostic metric is “normal.” The signs and symptoms do not fit any known disorder because they mix-and-match from the two or more that are contributing and they add new symptoms as a result of synergistic harm. This is not to say that there are no authentically new unsolved diseases. It is simply to say that if 1450 inborn errors of metabolism are the <2% tip of an iceberg whose main body is the interaction of partial defects in those 1450 disorders, then their interactions must be the prime goldmine to try to explain “new” disorders.
To put this another way, if something “new” can arise from interactions of everything old, the rate at which interactions appear mathematically must exceed the rate at which brand new non-interactive things arise.
More moderate cases are likely to fill the ranks of chronic fatigue, otherwise unexplained epilepsy, psychiatric disorders, developmental disorders, and so on.
Taking It Back to Wilson’s Disease
Now let’s take this back to Wilson’s disease. Let’s imagine someone is heterozygous for Wilson’s and a fatty acid oxidation disorder.
This hypothetical person shows no copper-related symptoms until they start running marathons on an MCT oil-supplemented keto diet while practicing intermittent fasting. Then, they start developing muscle cramps and hand tremors and become more irritable, their liver enzymes go up, and their serum copper drops. They figure they have copper deficiency, so supplement. This causes slurred speech or a psychotic break. They withdraw the copper, things start to normalize. They start eating regular carbohydrate-inclusive meals. The muscle cramps subside. Mood improves, hand tremors disappear, and labs all go back to normal.
What happened?
In this hypothetical case, the person was heterozygous for a fatty acid oxidation disorder. They did not have the classical presentation, but muscle cramps emerged as a minor symptom and their ATP levels crashed when they put maximal stress on fatty acid oxidation. Their copper metabolism was only half-dysregulated before this, but became fully dysregulated once the drop in ATP combined with the defective copper-transporting ATPase. As a result of the failure to distribute copper properly, copper supplementation made the neurological problems worse by making free copper accumulate in the nervous system. Removing the stresses on fatty acid oxidation and the problematic copper supplement made things return to normal.
This is just one example, and there could be tens of thousands more if we put our imagination to work.
Rare Metabolic Disease Genes Require Whole Genome Sequencing to Analyze
The rare disease genes we are discussing in this article requires whole exome or whole genome sequencing to analyze, and whole genome sequencing is superior.
Most of what shows up in common genetic analyses such as 23andMe and Ancestry are common single nucleotide polymorphisms. A nucleotide is a single “letter” in the “language” of genetic information, and a single nucleotide polymorphism or SNP is a one-letter difference in a genetic “word.” There are over 3 million SNPs. Some are of no consequence; some are important; the relevance of most is unclear. Since 2017, the method used by 23andMe captures about 640,000 of them. There are larger variations in the genetic code that are not captured or hardly captured at all in such tests, such as insertions, deletions, and duplications. These all vary in frequency from extremely common to extremely rare.
However, the variations that cause full-blown inborn errors of metabolism tend to be rare. The fact that full-blown diseases from traditional homozygosity are often fatal or debilitating helps keep their prevalence low.
Services such as 23andMe and Ancestry use a technique known as SNP-chips to categorize people as having or not having specific genetic variations known as single nucleotide polymorphism. This technique relies on testing a large batch of genomes at once and examining how they cluster with one another toward one or another of the binary outcomes. As such, giving a call to one person relies on relating that person to everyone else in the batch. As a result of this, the accuracy is directly related to the proportion of the people in the batch who have one or the other outcome. Since rare disease genes have very lopsided outcomes — most people don’t have the disease genotype — the accuracy falls.
Specifically, the accuracy on every metric is >99% for most SNPs tested, but the accuracy falls to 16% for variants with a frequency below 0.001%.
If the SNP-chip service includes a rare disease gene in an official report, they have likely taken measures to shore up the accuracy, such as using multiple differently designed probes for the same variant. However, any analysis of the raw data for such genes must be assumed to be 84% inaccurate.
Whole exome and genome sequencing use a far more accurate system of sequencing that “reads” pieces of a fragmented genome, called next-generation sequencing. The longer the fragments used, the more accurate the results. Accuracy of the raw data can exceed 99.99% with some platforms, and multiple reads of the genome can remove errors. For example, Dante Labs claims to use 99.9% accuracy to read the genome 30 times, where 30 re-reads would enable identification and correction of points with conflicting reads.
Whole exome sequencing covers 2% of the genome, and whole genome sequencing covers 90% of the genome. The exome is all of the exons, which are the portions of DNA that code for specific amino acids included in final proteins. However, this leaves out a lot of DNA that can impact how the gene is regulated and expressed. A 2018 meta-analysis found that the clinical utility of the two methods was not statistically significantly different, but the point estimate for the clinical utility score was 58% higher for whole genome sequencing than whole exome sequencing.
Why Most Genetic Information Is Noise
Most nutrigenomic approaches focus on common polymorphisms such as MTHFR rather than rare disease genes.
This is ignoring the most important signals to focus on the noise.
Consider this several ways:
The Pareto principle or 80/20 rule suggests that 80% of consequences come from 20% of causes in many cases. Given the commonness of this principle, we should be primed to look for the disproportionate results we can gain by focusing on the small number of things that are most important.
If 23andMe captures 640,000 polymorphisms and fails to capture the 2-12 rare disease genes, and fails to capture the 1-6 rare disease genes that are in the operating system rather than the apps, this is likely to be far more skewed than 80/20. It is more like the 0.001% of genes generating the 99.999% of results.
Common polymorphisms by definition on average produce the average, because they are defined by their deviation from normal. Rare disease genes on average produce a rare disease, because they are defined by their pathogenicity. Natural selection minimizes the presence of harmful genes, which makes the rarity of a gene correlate to its severity. Thus, the biggest problems have to be the few rare problems, not the many common variations.
Finally, consider this metaphor.
Imagine you are held captive and given 5 minutes to eat. The only available food is 1000 meal replacement drinks, each with a label with the nutrition facts, none of which are nutritionally balanced. 3 bottles are poison at half the dose that would kill a child and are marked with a red X on the nutrition label. What would you spend your five minutes on? Figuring out how to make a nutritionally balanced meal or avoiding the poison?
Clearly, your priority should avoiding the poison.
This focus on problems and “poison” may seem morbid, but consider that if we find the nutritionally actionable problems they become opportunities.
For example, I have a rare mutation in ACAD9 with a frequency of 0.08%. In its most severe form, it is a guaranteed death sentence in infancy. High-dose riboflavin, however, brings the fatality rate down to 10%.
For me, this gene represents an opportunity to impact my health with riboflavin. Indeed, 75 milligrams per day nearly immediately abolished my seasonal allergies and led to months of profound psychological benefits.
The High-Hanging-Fruit Unlock for Optimizing Wellness, Performance, and Longevity
To see why this is clearly the high-hanging-fruit unlock for optimizing wellness, performance, and longevity, let’s consider the collection of thiamin-responsive disorders, which include defects in thiamin transporters, the metabolism of thiamin, and in thiamin-dependent enzymes.
No single nutrient is as disproportionately involved in carbohydrate metabolism as thiamin.
If someone is heterozygous for one of these mutations, half of their carbohydrate metabolism will work normally, while the other half only works in the presence of 500-2000 milligrams of thiamin per day.
Consider an athlete who has optimized every last drop of training, nutrition, and recovery. This, with her strength, insanely hard work, and determination, led her to bring home the silver. If her carbohydrate metabolism was limited by heterozygosity for one of these mutations, would it not be the case that 500-2000 milligrams of thiamin would bring her home the gold?
Now consider someone who wants to live to 100.
Diabetes is found in 11.3% of Americans, and pre-diabetes brings this to 40.4% of Americans. Diabetes is the 7th leading cause of death, and its commonness makes it a primary obstacle to reaching centenarian status.
While addressing the low-hanging fruit by eating a healthy diet, maintaining a healthy body composition, and staying active, is what everyone should be doing to prevent diabetes, many people with mutations in the above genes will need 500-2000 milligrams per day of thiamin to optimize their carbohydrate metabolism and thus to prevent diabetes.
When we consider the 700-800 different nutritionally actionable rare disease genes in energy metabolism, and the likelihood that each person carries one to six of them, it simply must be the case that optimizing for wellness, performance, and longevity requires identifying the most important bugs in the operating system and debugging them.
Why Whole Genome Sequencing Isn’t Enough
While whole genome sequencing is necessary for debugging the OS, it is not even close to sufficient.
The standard output of whole genome sequencing is dozens of reports that each list out dozens of mutations.
There is no attempt to rank the mutations in importance, let alone to identify a small number that are most important.
There is no attempt to determine which mutations are at the deepest level of root cause.
There is no attempt to filter the mutations by actionability.
There is no attempt to vet the predictions made by those mutations according to biochemical evidence from lab tests showing what is actually happening in a person’s metabolism.
How to Debug the OS
I have developed a systematic approach to identifying the one to six most important glitches in each person’s operating system:
Rank rare disease genes according to their likely severity, their impact on energy metabolism, and their actionability.
Use biochemical labwork to determine which mutations appear to be actively limiting a person’s metabolism.
Develop a nutritional action plan around the most important bottlenecks.
Use the person’s response to the action plan to confirm, refute, or revise the model of their most important mutations. Use these revisions to finalize the plan.
If you want me to debug your OS for you, sign up for a call at bioopthealth.com, to see if my new Biochemical Optimization Program is right for you.
Subscribe to Harnessing the Power of Nutrients
Scientific expertise blended with out-of-the-box thinking for new practical ideas you can use to help yourself on your journey to vibrant health, by Chris Masterjohn, PhD.








That first paragraph about "basic things' should be pinned to every wall!
THANKS
Your comment about central nervous system sensitivity struck me. For the first 10 years of my triathlon hobby, I could place top10 in any sprint to mid distance race I entered (sprint/olympic). The latter 5 years, I can barely finish a sprint (16 miles usually) without severe cramping. I'm not sure what changed or if I have adrenal/CNS fatigue. I am a data scientist that studies biometrics. I'm curious if you have advice for me as to where I can start looking.